Il paziente che mi siede davanti con la sua scatola di ramipril, o di valsartan, quasi sempre a un certo punto me lo chiede: «Ma dottore, questa è solo per la pressione, vero?». È una domanda ragionevole, ed è anche il punto in cui di solito mi fermo un momento. Perché la verità è che abbassare la pressione è, tra tutte le cose che quella pillola fa, forse la meno interessante.
Per mezzo secolo abbiamo raccontato l'ipertensione come un problema idraulico: troppa spinta dentro i tubi, e questi farmaci come qualcosa che allenta la spinta. È una storia comoda e non è falsa — la pressione conta, eccome. Ma non è il motivo per cui i bloccanti del sistema renina-angiotensina stanno alla base di quasi ogni terapia che prescrivo. Il motivo è che l'ormone che questi farmaci disinnescano, l'angiotensina II, non è soltanto un vasocostrittore: è un ormone di rimodellamento, un segnale che dice al cuore di ispessirsi, al rene di logorarsi, alle arterie di irrigidirsi e infiammarsi. Bloccarlo significa spegnere un programma, non solo abbassare un numero.
Questo articolo prova a spiegare, meccanismo per meccanismo, perché ACE-inibitori e sartani sono il cardine — e, con la stessa onestà, dove finiscono i loro poteri. Perché un cardine non è una panacea. È pensato per i pazienti, non per i colleghi, ma il rigore è lo stesso che pretenderei da un collega.
Il sistema che regola la pressione (e molto altro)
Immaginiamo una catena di comando. All'inizio ci sono alcune cellule specializzate del rene, le cellule iuxtaglomerulari, sentinelle affacciate sui vasi che entrano nel glomerulo. Quando percepiscono che arriva meno sangue, o meno sale, o quando il sistema nervoso simpatico le sprona, rilasciano un enzima: la renina. È l'interruttore a monte di tutto il sistema — il passo che decide quanto forte girerà l'intera macchina.
La renina taglia una proteina prodotta dal fegato, l'angiotensinogeno, e ne ricava l'angiotensina I, una molecola quasi inerte. È il passaggio successivo a essere decisivo: un enzima chiamato ACE — enzima di conversione dell'angiotensina, presente soprattutto sull'endotelio dei polmoni ma anche nei tessuti — trasforma l'angiotensina I in angiotensina II, l'ormone effettore. L'angiotensina II, a sua volta, agisce su un recettore chiamato AT1 e ordina al surrene di liberare aldosterone, l'ormone che trattiene sodio e acqua.
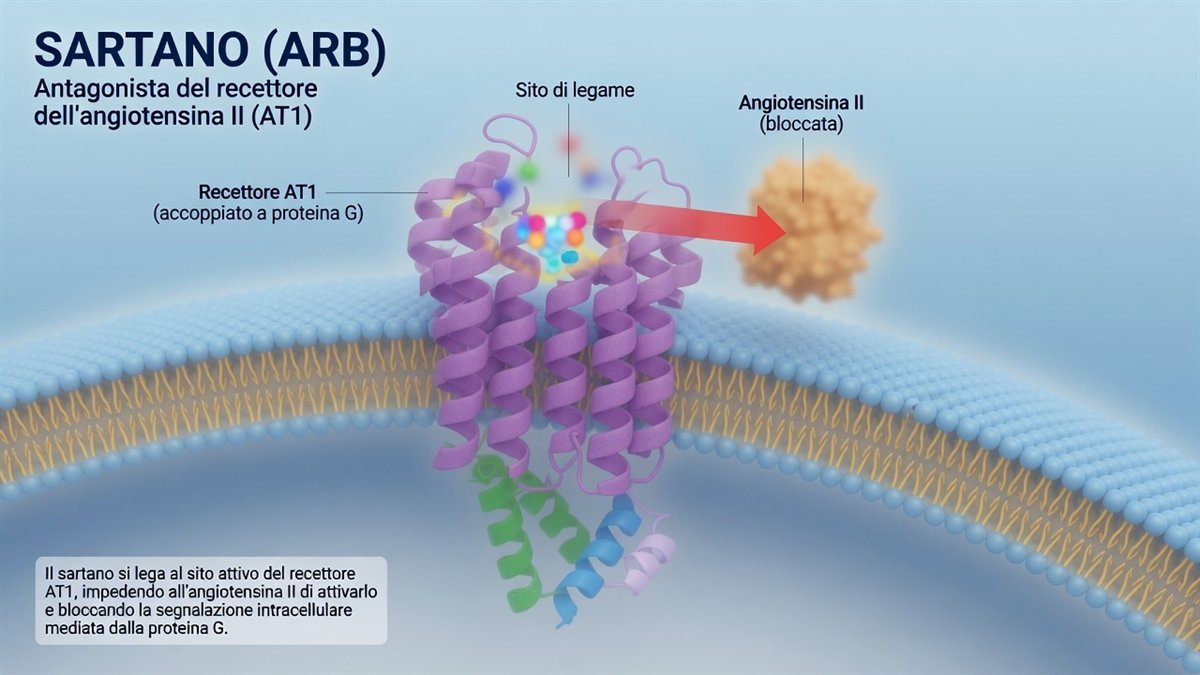
È qui che i due farmaci entrano in scena, in due punti diversi della stessa catena. L'ACE-inibitore blocca l'enzima ACE, a monte: meno angiotensina II viene prodotta. Il sartano lascia lavorare l'enzima ma blocca il recettore AT1, a valle: l'angiotensina II c'è, ma non trova la porta aperta. Due strategie, un solo obiettivo — togliere all'angiotensina II la sua voce.

Fin qui la fisiologia da manuale. Ma la parte che cambia davvero il modo di ragionare è un'altra, e riguarda che cosa faccia questo ormone una volta che ha la voce.
L'angiotensina II non è solo un vasocostrittore
Se l'angiotensina II si limitasse a stringere le arterie, bloccarla equivarrebbe a qualunque altro modo di abbassare la pressione. Non è così. Attraverso il recettore AT1, questo ormone accende almeno quattro programmi diversi di danno d'organo, e la vasocostrizione è solo il più visibile.

Il cuore che si irrigidisce e cicatrizza
L'angiotensina II parla direttamente ai cardiomiociti — le cellule muscolari del cuore — e dice loro di crescere. È un'ipertrofia che avviene in parte indipendentemente dal carico di pressione, per stimolo diretto. Ma il capitolo più insidioso riguarda i fibroblasti, le cellule dell'impalcatura: l'angiotensina II li spinge a proliferare e ad attivare una molecola chiave, il TGF-beta, che ordina la deposizione di collagene. Il risultato è la fibrosi — tessuto cicatriziale che si infiltra tra le fibre muscolari, tra un vaso e l'altro.
Un cuore fibrotico è un cuore che si irrigidisce: fatica a rilassarsi e a riempirsi (quella che chiamiamo disfunzione diastolica), e la cicatrice diventa un terreno elettricamente disomogeneo, il substrato su cui nascono le aritmie. Attenzione, però: la fibrosi e l'ipertrofia non sono opera esclusiva dell'angiotensina II — concorrono il carico pressorio, l'aldosterone, le catecolamine. Bloccare il sistema renina-angiotensina non cancella la cicatrice, ma contribuisce a ridurla, spesso in misura sproporzionata rispetto al solo effetto sulla pressione.
La ruggine che spegne l'ossido nitrico
Il secondo programma è più sottile e si gioca dentro la parete dei vasi. L'angiotensina II attiva un enzima, la NADPH-ossidasi, che produce anione superossido: molecole reattive dell'ossigeno, una sorta di ruggine biochimica. Questo superossido reagisce con l'ossido nitrico — il gas che l'endotelio sano produce per far dilatare e rilassare le arterie — e lo neutralizza.
Meno ossido nitrico disponibile significa un'arteria che si dilata peggio, più rigida, più reattiva: è la disfunzione endoteliale, il primo passo, silenzioso, verso l'aterosclerosi. Ed è anche il perno del ragionamento sul beneficio «oltre la pressione»: se questi farmaci restituiscono ossido nitrico e tolgono ruggine, dovrebbero proteggere il vaso al di là del semplice calo pressorio. È un'idea affascinante — e, come vedremo, va maneggiata con prudenza, perché la clinica l'ha confermata solo in parte.
Infiammazione, coaguli e ritenzione di sale
Restano gli altri due bracci. L'angiotensina II accende un fattore di trascrizione, l'NF-kB, che induce la parete del vaso a esporre molecole di adesione: veri e propri ganci a cui i globuli bianchi si attaccano per infiltrarsi nella parete e alimentare la placca. Nello stesso tempo aumenta la produzione di PAI-1, una molecola che frena la dissoluzione dei coaguli — spostando l'equilibrio verso uno stato pro-trombotico.
Infine il braccio più classico: l'aldosterone, stimolato dall'angiotensina II, fa riassorbire sodio e acqua nel rene, con espansione del volume e più lavoro per il cuore. Ma l'aldosterone non si limita al rene — ha effetti diretti pro-fibrotici su cuore e vasi, il che spiega perché in alcuni pazienti selezionati aggiungiamo un antagonista specifico. Quattro programmi, un solo mittente. Bloccare il mittente li spegne tutti insieme.
Due modi di spegnere lo stesso interruttore
A questo punto la domanda diventa pratica: se ACE-inibitore e sartano agiscono sulla stessa catena, che differenza c'è? La differenza è il punto in cui la interrompono, e ha una conseguenza concreta.
L'ACE-inibitore blocca l'enzima ACE. Ma quell'enzima ha un secondo mestiere: degradare la bradichinina, una molecola vasodilatatrice. Bloccando l'enzima, la bradichinina si accumula. Da un lato è un bene — contribuisce alla vasodilatazione e alla produzione di ossido nitrico. Dall'altro è la causa dell'effetto collaterale più noto: la tosse secca, stizzosa, che compare in una quota di pazienti (le stime pratiche parlano del 5-15%, con punte fino al 35% in alcune casistiche, più frequente nelle donne). E, molto più raramente, la stessa bradichinina è implicata nell'angioedema, un gonfiore che può essere serio.
Il sartano, invece, blocca direttamente il recettore AT1, a valle: non tocca la bradichinina. Niente accumulo, niente tosse, e un rischio di angioedema molto più basso — anche se, va detto con onestà, non nullo. C'è di più: bloccando il recettore, l'angiotensina II resta in circolo e può stimolare un altro recettore, l'AT2, che in laboratorio sembra mediare segnali opposti e protettivi. È un'ipotesi meccanicistica elegante, ma per ora prevalentemente da studi sperimentali: non c'è la prova che renda i sartani clinicamente superiori agli ACE-inibitori.
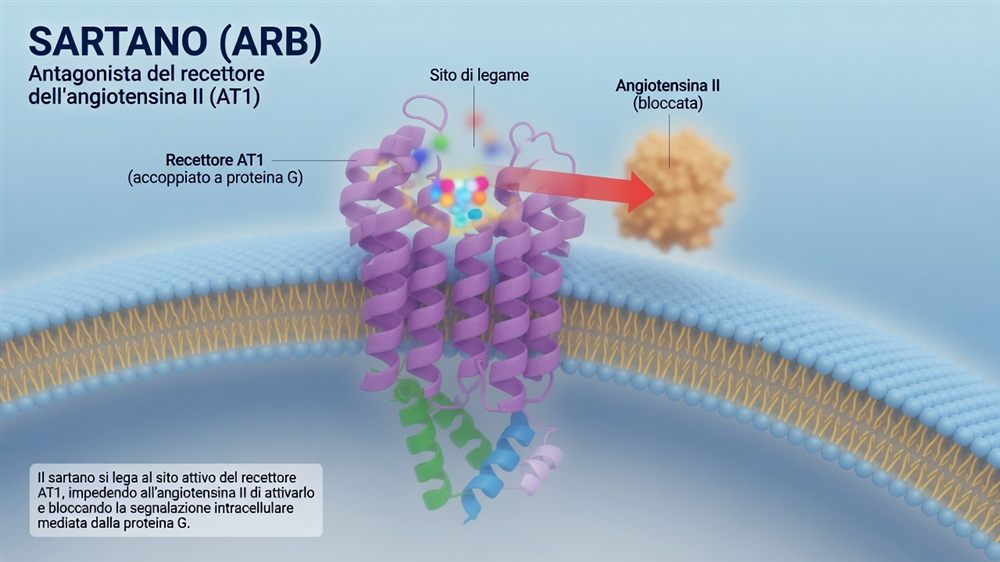
L'illustrazione qui sopra rende bene l'idea: il sartano si incastra nel recettore AT1, immerso nella membrana della cellula, e occupa il posto dove si sarebbe legata l'angiotensina II. L'ormone resta fuori, e il segnale che avrebbe scatenato — attraverso la proteina G accoppiata al recettore — non parte.

Ed è il punto che voglio far arrivare senza equivoci: sugli esiti — infarti, ictus, morte cardiovascolare — ACE-inibitori e sartani si equivalgono. Lo ha dimostrato in modo netto lo studio ONTARGET (New England Journal of Medicine, 2008), su oltre 25.000 pazienti ad alto rischio: il telmisartan non è risultato inferiore al ramipril. La scelta tra i due, quindi, non è una gara di efficacia. È una questione di tollerabilità — e la tosse da ACE-inibitore è, nella pratica di tutti i giorni, il motivo numero uno per cui passo un paziente al sartano.
Le prove sull'uomo, organo per organo
La fisiologia è convincente, ma da sola non basta. Quello che rende questi farmaci il cardine è che il beneficio d'organo è stato misurato, sull'uomo, in decine di studi controllati. Vale la pena guardarli organo per organo, perché è lì che la teoria diventa medicina.
Il cuore: regressione, non solo controllo
Lo studio che ha cambiato il modo di pensare si chiama LIFE (Lancet, 2002): oltre 9.000 ipertesi che avevano già un'ipertrofia del ventricolo sinistro all'elettrocardiogramma. Confrontava un regime a base di losartan con uno a base di atenololo, un beta-bloccante. A parità di calo pressorio — questo è il punto — il losartan riduceva l'endpoint cardiovascolare composito del 13%, trainato da una riduzione dell'ictus di circa il 25%, e faceva regredire di più la massa del ventricolo. Chi vedeva il proprio cuore «sgonfiarsi» aveva, indipendentemente, meno eventi.
Un chiarimento, per non gonfiare il dato: LIFE confrontava il losartan con l'atenololo, oggi non più di prima scelta nell'ipertensione non complicata. Non dimostra che «il losartan batte tutto»; dimostra che far regredire l'ipertrofia, e non solo abbassare la pressione, ha un valore.
Il capitolo più solido è però quello dello scompenso cardiaco e del post-infarto, dove il concetto di rimodellamento inverso — un cuore dilatato e sofferente che recupera forma e funzione — è nato proprio con questi farmaci. Lo storico CONSENSUS (1987) mostrò che nell'insufficienza cardiaca più grave l'enalapril riduceva la mortalità di circa il 40%; lo SOLVD la confermò nelle forme meno avanzate. Dopo un infarto con cuore indebolito, studi come SAVE, AIRE e TRACE dimostrarono che partire subito con un ACE-inibitore riduceva la mortalità e frenava la discesa verso lo scompenso. E i sartani? Nel post-infarto (studio VALIANT, 2003) il valsartan è risultato non inferiore al captopril — non superiore: la formula corretta è questa — mentre nello scompenso i programmi Val-HeFT e CHARM hanno confermato il beneficio di classe, con il candesartan utile anche in chi l'ACE-inibitore non lo tollerava.
Nel cuore, questi farmaci non si limitano a scaricare la pressione: interrompono un programma di rimodellamento maladattativo. È la differenza tra togliere peso a una trave e impedire che la trave si deformi.
La fibrillazione atriale: una promessa da maneggiare con cura
Qui devo essere particolarmente preciso, perché è il punto su cui si dicono più imprecisioni. Se l'angiotensina II fibrotizza anche l'atrio, allora bloccarla dovrebbe prevenire la fibrillazione atriale — l'aritmia più comune, che nasce proprio su un atrio dilatato e cicatrizzato. L'idea si chiama «terapia a monte», upstream therapy, e in parte funziona. Ma solo in parte, e la distinzione è tutto.

Dove funziona: nella prevenzione primaria, cioè nel cuore che ha già il substrato ma non ha ancora sviluppato l'aritmia. Nel sottostudio di LIFE (JACC, 2005), tra gli ipertesi con ipertrofia ventricolare il losartan riduceva la fibrillazione atriale di nuova insorgenza di un terzo rispetto all'atenololo (rischio relativo 0,67), a parità di pressione. Lo stesso vale, per estensione, nello scompenso e dopo l'infarto: agendo sul rimodellamento atriale prima che l'aritmia si instauri.
Dove non funziona: nella prevenzione secondaria, cioè per evitare le recidive quando la fibrillazione è già comparsa, e nella fibrillazione isolata di un cuore strutturalmente sano. Qui gli studi dedicati — GISSI-AF con il valsartan, ANTIPAF con l'olmesartan — sono stati negativi. Non riducono le recidive, non «sciolgono» un'aritmia già stabilita. Chi promette che un sartano «previene la fibrillazione» in senso generale sta dicendo qualcosa che i dati non sostengono.
Il rene: il glomerulo a due rubinetti
È forse il territorio dove il beneficio d'organo è più elegante e meglio dimostrato, e per capirlo serve un'immagine. Il glomerulo — l'unità filtrante del rene — è come un lavandino con due rubinetti: un vaso che porta il sangue dentro (l'arteriola afferente) e uno che lo porta fuori (l'arteriola efferente). La pressione con cui il sangue viene filtrato dipende dall'equilibrio tra i due.
L'angiotensina II ha una preferenza: costringe soprattutto l'arteriola in uscita, l'efferente. È come strozzare lo scarico — la pressione dentro il glomerulo sale, la filtrazione si fa più violenta. Nel breve periodo il rene sembra funzionare bene, ma quella iperfiltrazione è un logorio: fa passare proteine nelle urine e, anno dopo anno, sfianca il filtro. I bloccanti del sistema renina-angiotensina fanno l'opposto: dilatano l'efferente, riaprono lo scarico, e la pressione dentro il glomerulo scende.

Ecco perché — e qui sta la parte controintuitiva — questi farmaci proteggono il rene in un modo che gli altri antipertensivi non hanno. Lo dimostra un vero e proprio esperimento naturale, lo studio IDNT (2001): in pazienti con diabete e danno renale, l'irbesartan e l'amlodipina abbassavano la pressione allo stesso modo, ma solo l'irbesartan proteggeva il rene, riducendo l'endpoint renale del 23% rispetto all'amlodipina. Stessa pressione, destino diverso: la differenza si giocava dentro il glomerulo.
La lista degli studi che hanno costruito questa certezza è lunga, ed è importante non confondere i risultati tra loro — ogni cifra vale per la sua popolazione e per il suo specifico traguardo.

Lo studio fondativo (Lewis e i colleghi del Collaborative Study Group, 1993) riguardava il diabete di tipo 1 con proteinuria: il captopril riduceva del 48% il rischio di raddoppio della creatinina — un marcatore di rene che peggiora. In RENAAL (2001), su pazienti con diabete di tipo 2, il losartan riduceva del 28% il rischio di arrivare alla dialisi. IRMA-2, sempre nel 2001, mostrava che nelle fasi precoci, quando nelle urine compaiono solo tracce di proteine, l'irbesartan a dose piena riduceva di circa il 70% la progressione verso la proteinuria conclamata. E il beneficio non si ferma ai diabetici: in REIN (1997) il ramipril rallentava il declino del rene anche nelle nefropatie non diabetiche, e in AASK (2002), in una popolazione afroamericana con danno renale da ipertensione, si è mostrato superiore all'amlodipina.
Un dettaglio che spesso spaventa il paziente, e che vale la pena spiegare. Nelle prime settimane di terapia la creatinina — l'esame del sangue che misura la funzione del rene — può salire un poco. Non è, entro certi limiti, un effetto avverso: è il segno emodinamico che il farmaco sta lavorando sul glomerulo, esattamente come deve. Un rialzo fino a circa il 30%, che poi si stabilizza, è atteso e persino desiderabile.
L'endotelio e la placca: il terreno più incerto
Arriviamo al capitolo che richiede più equilibrio, quello vascolare. La logica è quella dell'ossido nitrico di cui parlavamo: se questi farmaci migliorano la funzione dell'endotelio e riducono lo stress ossidativo, dovrebbero proteggere le arterie dall'aterosclerosi oltre e in aggiunta al calo pressorio. Alcuni grandi studi lo hanno suggerito. HOPE (2000) mostrò che il ramipril, in pazienti ad alto rischio ma senza scompenso noto, riduceva del 22% gli eventi cardiovascolari maggiori, con un calo di pressione in ambulatorio sorprendentemente modesto. EUROPA (2003) confermò il beneficio del perindopril nella coronaropatia stabile.
Ma un buon articolo non può fermarsi qui, e nemmeno un buon medico. Perché lo stesso studio HOPE, quando la pressione fu misurata nelle 24 ore in un piccolo sottogruppo, mostrò un calo notturno molto maggiore di quello registrato in ambulatorio: buona parte del beneficio, insomma, potrebbe essere semplicemente pressione abbassata meglio, non un effetto misterioso «oltre la pressione». E soprattutto c'è PEACE (2004): in pazienti con coronaropatia stabile ma ben curati per il resto — statine, rivascolarizzazione, beta-bloccanti — l'aggiunta di un ACE-inibitore non ha aggiunto nulla. Uno studio negativo, che va citato accanto agli altri per onestà.
La formula corretta è «oltre e in aggiunta al controllo pressorio», mai «indipendentemente dalla pressione». L'effetto extra c'è, ma è di entità modesta e dipende da chi hai davanti: enorme in chi ha rene o cuore già sofferenti, sfumato in chi è già ben protetto.
E vale un ultimo scrupolo di linguaggio: questi farmaci non «sciolgono» la placca né «guariscono» l'arteria. Semmai contribuiscono a stabilizzarla e a rallentare il processo. Il resto è entusiasmo, e l'entusiasmo, in medicina, è quasi sempre l'anticamera dell'imprecisione.
Perché sono il cardine — e cosa non sono
Torniamo alla domanda del paziente con la scatola in mano. Se il beneficio «oltre la pressione» è reale ma misurato, perché questi farmaci stanno alla base di quasi ogni terapia? La risposta è duplice, e nessuno dei due motivi è «perché abbassano la pressione meglio degli altri».
Il primo motivo è strutturale. Le linee guida più recenti — la Società Europea di Cardiologia, nel documento del 2024 — collocano ACE-inibitori e sartani tra i farmaci di prima linea, e soprattutto li rendono lo scheletro delle combinazioni preferite: un bloccante del sistema renina-angiotensina insieme a un calcio-antagonista, oppure insieme a un diuretico, idealmente in una singola compressa che unisce i due principi. Non è un caso: si combinano bene con quasi tutto, con effetti che si sommano e tollerabilità che spesso migliora (il bloccante del RAAS, per esempio, tende a smorzare i gonfiori alle caviglie dei calcio-antagonisti).
Il secondo motivo è qualitativo, ed è tutto quello che abbiamo detto finora. Quando davanti ho un cuore ipertrofico, uno scompenso, un rene che perde proteine, un diabetico — cioè un organo che sta già soffrendo — questi farmaci non abbassano soltanto un numero: proteggono quell'organo. È questa la loro centralità. A parità di calo pressorio, in un iperteso non complicato, un diuretico può fare altrettanto bene (lo mostrò ALLHAT, nel 2002). Ma la medicina non tratta ipertesi astratti: tratta persone con storie e organi bersaglio, e lì la scelta del bloccante del RAAS smette di essere una tra tante e diventa quasi obbligata.
Vale la pena rendere concreto questo «quasi obbligata». Ci sono situazioni in cui, davanti a un paziente, un ACE-inibitore o un sartano diventano la spina dorsale della terapia quasi a prescindere dal valore di pressione, perché è lì che il beneficio d'organo è dimostrato:
- il diabetico con anche solo tracce di proteine nelle urine — la microalbuminuria: è il segnale precoce che il rene sta soffrendo, e questi farmaci ne sono da decenni il presidio di riferimento per rallentarne il logorio (oggi affiancati, non sostituiti, da altri nefroprotettori più recenti);
- chi ha una malattia renale cronica che perde proteine, diabetico o no;
- chi ha uno scompenso cardiaco a funzione ridotta, dove riducono i ricoveri e la mortalità;
- chi ha avuto un infarto con il cuore indebolito, dove frenano il rimodellamento;
- chi ha un'ipertrofia del ventricolo sinistro, il cuore ispessito che chiede di regredire.
In tutti questi casi la domanda non è più «quale antipertensivo», ma «come costruisco la terapia attorno al bloccante del RAAS». È questo che significa, nella pratica, essere il cardine.
Le tre regole intoccabili
Proprio perché sono ovunque, vale la pena fissare i pochi confini che non vanno mai varcati — e che un paziente informato può conoscere.
La prima: ACE-inibitore e sartano non si combinano mai tra loro. Fanno lo stesso mestiere, e raddoppiare il blocco non raddoppia la protezione: la peggiora. Lo studio ONTARGET, e nel rene lo studio VA NEPHRON-D, hanno mostrato che l'associazione dei due aumenta ipotensione, danno renale acuto e potassio alto senza alcun beneficio. Lo stesso vale per l'aggiunta di un terzo bloccante, l'aliskiren, nel paziente diabetico (studio ALTITUDE, interrotto per un eccesso di eventi). La protezione è di classe: non si potenzia moltiplicando i farmaci sullo stesso bersaglio.
La seconda: mai in gravidanza. Questi farmaci sono tossici per il feto, in particolare nel secondo e terzo trimestre, e vanno sospesi e sostituiti in ogni donna che programmi o inizi una gravidanza. È una controindicazione assoluta, non una cautela.
La terza: attenzione alla stenosi delle arterie renali quando è bilaterale. In quel caso il rene dipende proprio dall'angiotensina II per mantenere la propria pressione di filtrazione: toglierla di colpo può far precipitare la funzione. È una delle ragioni per cui quel controllo del sangue nelle prime settimane non è una formalità.
Vale la pena aggiungere una nota per non generare confusione: esiste un farmaco più recente, il sacubitril/valsartan, che unisce un sartano a una seconda molecola. È un pilastro dello scompenso cardiaco a funzione ridotta, non un farmaco per l'ipertensione comune, e la sua introduzione richiede regole precise (tra cui una pausa di almeno 36 ore da un eventuale ACE-inibitore, per evitare l'angioedema). È l'evoluzione della storia, non il suo inizio.
Cardine, non miracolo
Alla fine, quando il paziente mi richiede se «è solo per la pressione», la risposta più vera è: no, e questo è il bello, ma anche no, e questo è il limite. Non è solo per la pressione, perché quel farmaco sta parlando al cuore che vuole ispessirsi, al rene che vuole logorarsi, all'arteria che vuole irrigidirsi, e sta dicendo loro di fermarsi. Ma non è nemmeno un talismano: l'effetto oltre la pressione è reale e misurato, non illimitato; funziona benissimo dove c'è un organo da proteggere e molto meno dove non c'è; previene la fibrillazione atriale prima che nasca, non dopo; stabilizza la placca, non la scioglie.
È esattamente questa misura a renderlo il cardine. In medicina i farmaci che promettono tutto di solito mantengono poco. Questi promettono una cosa precisa — interrompere un programma di danno che parte da un unico ormone — e la mantengono, in modo dimostrato, negli organi giusti. Non è poco. È, probabilmente, il motivo per cui quella scatola finisce nella tasca di così tante persone.
L'angiotensina II è un ordine che il corpo dà a se stesso, e non sempre è un ordine saggio. Il compito di questi farmaci, in fondo, è semplice: interrompere la comunicazione prima che il messaggio faccia danno.
Fonti
- McMurray JJV et al. Telmisartan, ramipril, or both in patients at high risk for vascular events (ONTARGET). New England Journal of Medicine, 2008.
- Dahlöf B et al. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE). Lancet, 2002; e Wachtell K et al., sottostudio sulla fibrillazione atriale di nuova insorgenza, Journal of the American College of Cardiology, 2005.
- The CONSENSUS Trial Study Group. Effects of enalapril on mortality in severe congestive heart failure. New England Journal of Medicine, 1987; SOLVD Investigators, 1991.
- Pfeffer MA et al. Valsartan, captopril, or both in myocardial infarction complicated by heart failure, left ventricular dysfunction, or both (VALIANT). New England Journal of Medicine, 2003. Studi SAVE, AIRE, TRACE sul post-infarto.
- Lewis EJ et al. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy (Collaborative Study Group). New England Journal of Medicine, 1993.
- Brenner BM et al. (RENAAL) e Lewis EJ et al. (IDNT); Parving HH et al. (IRMA-2). New England Journal of Medicine, 2001.
- GISEN Group. Randomised placebo-controlled trial of effect of ramipril on decline in glomerular filtration rate in proteinuric non-diabetic nephropathy (REIN). Lancet, 1997; Wright JT et al. (AASK), JAMA, 2002.
- Yusuf S et al. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients (HOPE). New England Journal of Medicine, 2000; The EUROPA Investigators, Lancet, 2003; The PEACE Trial Investigators, New England Journal of Medicine, 2004.
- GISSI-AF Investigators, New England Journal of Medicine, 2009; Goette A et al. (ANTIPAF), Circulation: Arrhythmia and Electrophysiology, 2012.
- Fried LF et al. (VA NEPHRON-D), New England Journal of Medicine, 2013; Parving HH et al. (ALTITUDE), New England Journal of Medicine, 2012.
- McEvoy JW, Touyz RM et al. 2024 ESC Guidelines for the management of elevated blood pressure and hypertension. European Heart Journal, 2024.